









【成果のポイント】
・「QM/MM(量子力学/分子力学)法」注1)は、巨大で複雑な分子系の機能を高精度かつ高効率に扱うための中核的な分子シミュレーション手法として広く用いられています。しかし、「巨大分子系のどこまでを量子力学で精密に扱うべきか」という QM 領域の設定は、研究者の経験に依存しており、予測性や再現性の観点から長年の課題となっていました。
・本研究では、分子認識や化学反応に伴って生じる分子軌道や電荷分布の変化といった電子状態応答に着目し、これらが QM 領域を定めるための客観的かつ明確な物理指標となり得ることを示しました。この電子状態応答は、QM/MM 計算に先立って、ごく簡便な領域分割型の半経験(semi-empirical)注2)QM 計算を一度行うだけで、十分な精度で評価可能です。
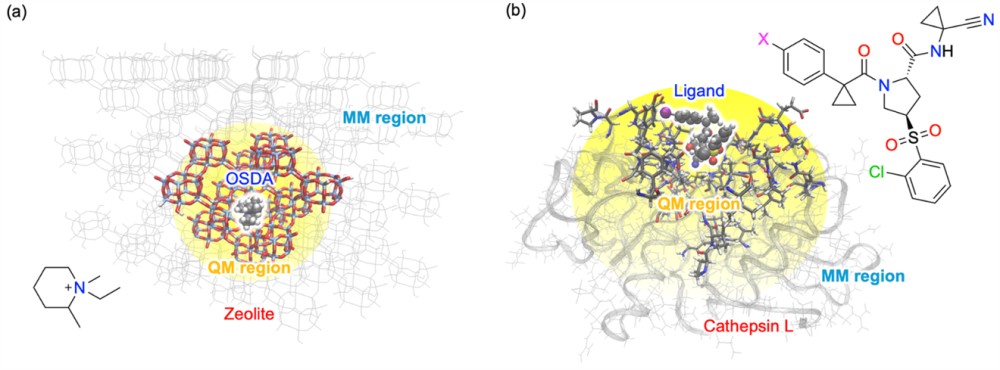
・提案手法は、ゼオライト–構造規定剤複合体や、ヒトカテプシン–阻害剤複合体といった、材料科学および生命科学の双方において重要な系を含む幅広いモデルで検証され、異なる QM 計算条件に対しても一貫して適用可能であることを確認しました。
・今後、本手法は、創薬や高機能材料開発における予測的分子設計を支える、実用的な計算プラットフォームとしての展開が期待されます。
【概 要】
中央大学理工学部応用化学科の森寛敏教授と、お茶の水女子大学の小澤二千夏(博士後期課程1年)・黒木菜保子助教らの研究グループは、分子シミュレーション手法 「QM/MM 法(量子力学/分子力学法)」において量子力学的に扱う領域を、電子状態変化に基づき客観的かつ自動的に定義する新しい設計原理を提案しました。
QM/MM 法は、化学反応や分子認識に直接関与する重要領域のみを量子力学で扱い、それ以外の領域を分子力学で簡略化して記述することで、材料中での化学反応・分子認識の理解や、創薬・生命科学に関わる酵素反応の解析など、巨大分子系を現実的な計算コストで解析できる強力な手法です。一方で、QM 領域と MM 領域の境界設定が研究者の経験や直感に依存してきたため、予測的な適用や再現が難しいという課題がありました。
本研究では、化学反応や分子認識に伴って生じる電子状態応答(分子軌道エネルギーの変化や電荷の再分配)に着目することで、この課題に新たな解決策を与えました。すなわち、全体系に対して領域分割型の半経験QM計算を一度だけ行い、そこで得られる電子状態応答を解析すれば、どの領域を量子力学的に扱うべきかを物理的根拠に基づいて迅速に判断できることを示しました。
実際に本設計原理を、ゼオライト–構造規定剤複合体や、ヒトカテプシン–阻害剤複合体といった、性質の異なる複数の系(無機材料や生体分子)に適用した結果、いずれの系においてもエネルギー評価は化学精度を維持し、QM/MM 計算が予測的に機能することが確認されました。本原理は特定の量子化学手法に依存しないため、密度汎関数法(DFT)注3)や非経験(ab initio)法注4)など、より高精度な計算手法への展開も可能です。
本成果は、QM/MM 法を「実験結果の説明手段」から、「分子機能や反応性を事前に予測・設計するための理論基盤」へと発展させるものです。今後は、本研究で確立した電子状態応答による設計原理を、機械学習や AI 技術と組み合わせ、さらには予測に基づく狙い撃ち実験へと応用することで、複雑な材料や反応系に対する予測科学の深化や設計自動化へとつながる可能性も期待されます。
本成果は、2025年12月23日(日本時間)付けで、材料科学・生命科学・化学・物理学などの先端分野に関わる、分野横断的かつ概念的なブレークスルーを示す研究成果を掲載する国際的総合学術誌 『Advanced Science』に Early View (オンライン先行版)として掲載されました。
【参考情報】
2025年12月23日に、本成果概要を中央大学理工学部応用化学科Webページで公開しています。
https://www.chuo-u.ac.jp/academics/faculties/science/departments/chemistry/news/2025/12/83653/
【研究者】
森 寛敏 中央大学 理工学部 教授 (応用化学科)
小澤 二千夏 お茶の水女子大学 大学院人間文化創成科学研究科 博士後期課程1年(理学専攻 化学・生物化学領域)
黒木 菜保子 お茶の水女子大学 基幹研究院 助教(自然科学系)
【掲載誌】
Ozawa Nichika, Kuroki Nahoko & Mori Hirotoshi*,
Ligand-Induced Electronic Response Enables Predictive QM/MM Simulations
Adv. Sci. 2025, published online (Early View).
https://advanced.onlinelibrary.wiley.com/doi/10.1002/advs.202519137 (Open Access)
【用語解説】
注1)QM/MM(量子力学/分子力学)法
酵素反応、薬剤とタンパク質の相互作用、材料中での化学反応など、巨大で複雑な分子系を扱う際に広く用いられている分子シミュレーション手法。量子力学(QM)と分子力学(MM)を組み合わせたハイブリッド型の計算手法である。分子の中で反応や結合に直接関与する重要な部分を QM で精密に計算し、それ以外の部分を古典的な MM で近似することで、計算精度と計算効率の両立を図る。一方で、どこまでを QM で扱い、どこから MM とするかという領域境界の決定は、従来は計算者の経験や専門的知識に基づいて慎重に定める必要があった。
注2)半経験(semi-empirical)法
量子力学に基づきつつ、実験データや高精度計算から得られた経験的パラメーターを取り入れることで、計算を簡略化した電子状態計算手法。計算精度は用いるパラメーターに依存するが、計算速度に優れており、大規模分子系や多数の構造を効率的に評価したい場合に適している。
注3)密度汎関数法(DFT)
分子や材料の性質を決定する電子の振る舞いを、電子密度に基づいて計算する量子化学手法。分子中のすべての電子の運動を厳密に追跡することは計算量の観点から困難であるが、DFT では電子の集まりとしての分布(電子密度)に着目することで、計算負担を抑えながら実用的な化学精度を得ることができる。そのため、分子構造の解析、反応エネルギー評価、材料の電子的性質の解析などに広く用いられており、現在の計算化学・材料科学における標準的な手法の一つである。
注4)非経験(ab initio)法
実験データや経験的なパラメーターに依存せず、量子力学の基本原理のみに基づいて分子の電子状態を計算する方法。「ab initio」はラテン語で「最初から」を意味する。電子同士の相互作用(電子相関)をできるだけ厳密に取り扱うため、高い精度で分子の性質を予測できる一方、計算負担が大きくなる。小分子系や反応機構の詳細解析など、高精度な理論的理解が求められる場面で主に用いられる。
詳細は、大学ホームページの「プレスリリース」をご覧ください。
また、ご興味をお持ちの方は下記もご覧ください。